ComputeIncoherentDOS v1#
Summary#
Calculates the neutron weighted generalised phonon density of states in the incoherent approximation from a measured powder INS MatrixWorkspace
Properties#
Name |
Direction |
Type |
Default |
Description |
|---|---|---|---|---|
InputWorkspace |
Input |
Mandatory |
Input MatrixWorkspace containing the reduced inelastic neutron spectrum in (Q,E) or (2theta,E) space. |
|
Temperature |
Input |
number |
300 |
Sample temperature in Kelvin. |
MeanSquareDisplacement |
Input |
number |
0 |
Average mean square displacement in Angstrom^2. |
QSumRange |
Input |
string |
0,Qmax |
Range in Q (in Angstroms^-1) to sum data over. |
EnergyBinning |
Input |
string |
0,Emax/50,Emax*0.9 |
Energy binning parameters [Emin, Emax] or [Emin, Estep, Emax] in meV. |
Wavenumbers |
Input |
boolean |
False |
Should the output be in Wavenumbers (cm^-1)? |
StatesPerEnergy |
Input |
boolean |
False |
Should the output be in states per unit energy rather than mb/sr/fu/energy? (Only for pure elements, need to set the sample material information) |
OutputWorkspace |
Output |
Mandatory |
Output workspace name. |
|
TwoThetaSumRange |
Input |
string |
Twothetamin, Twothetamax |
Range in 2theta (in degrees) to sum data over. |
Description#
Computes the phonon density of states from an inelastic neutron scattering measurement of a powder or polycrystalline sample, assuming that all scattering is incoherent, using the formula for the 1-phonon incoherent scattering function [1]:
where the term in square brackets is the neutron weighted density of states which is calculated by this algorithm, and \(g_k(E)\) is the partial density of states for each component (element or isotope) \(k\) in the material. \(m_k\) is the relative atomic mass of the component.
The average Debye-Waller factor \(\exp\left(-2\bar{W}(Q)\right)\) is calculated using an average mean-square displacement \(\langle u^2 \rangle\), using \(W=Q^2\langle u^2\rangle/2\).
\(\langle u^2 \rangle\) is also called the isotropic atomic displacement parameter \(U_{\mathrm{iso}}\) in Rietveld refinement programs like GSAS. There is also another, related, type of atomic displacement parameter used in programs like FullProf called \(B_{\mathrm{iso}}\) : \(B_{\mathrm{iso}}=8\pi^2U_{\mathrm{iso}}\) [2].
The algorithm accepts both \(S(Q,E)\) workspaces as well as
\(S(2\theta,E)\) workspaces. In the latter case \(Q\) values are
calculated from \(2\theta\) and \(E\) before applying the formula
above. Note, that QSumRange is only applicable with \(S(Q,E)\) while
TwoThetaSumRange works only for \(S(2\theta,E)\).
If the data has been normalised to a Vanadium standard measurement, the output of this algorithm is the neutron weighted density of states in milibarns/steradians per formula unit per meV (or per cm^-1). If the sample material has been set and is found to be a pure element, then an additional option will be enabled to calculate the DOS in states per meV (states per cm^-1) by dividing by the scattering cross-section and multiplying by the relative atomic mass.
Restrictions on the Input Workspace#
The input workspace must have units of Momentum Transfer or Degrees and contain histogram data with common binning on all spectra.
Usage#
Note
To run these usage examples please first download the usage data, and add these to your path. In Mantid this is done using Manage User Directories.
ISIS Example
The following code will run a reduction on a MARI (ISIS) dataset and apply the algorithm to the reduced data. The datafiles (runs 21334, 21335, 21347) and map file ‘mari_res2013.map’ should be in your path. Run number 21335 is a measurement of a large Aluminium sample from the neutron training course.
from Direct import DirectEnergyConversion
from mantid.simpleapi import *
rd = DirectEnergyConversion.DirectEnergyConversion('MARI')
ws = rd.convert_to_energy(21334, 21335, 60, [-55,0.05,55], 'mari_res2013.map',
monovan_run=21347, sample_mass=106.4, sample_rmm=27, monovan_mapfile='mari_res2013.map')
ws_sqw = SofQW3(ws, [0,0.1,12], 'Direct', 60)
SetSampleMaterial(ws_sqw,'Al')
ws_dos = ComputeIncoherentDOS(ws_sqw, Temperature=5, StatesPerEnergy=True)
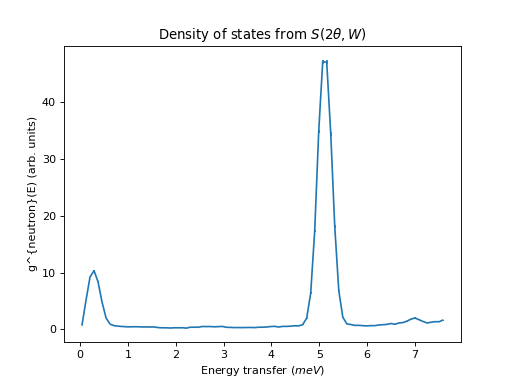
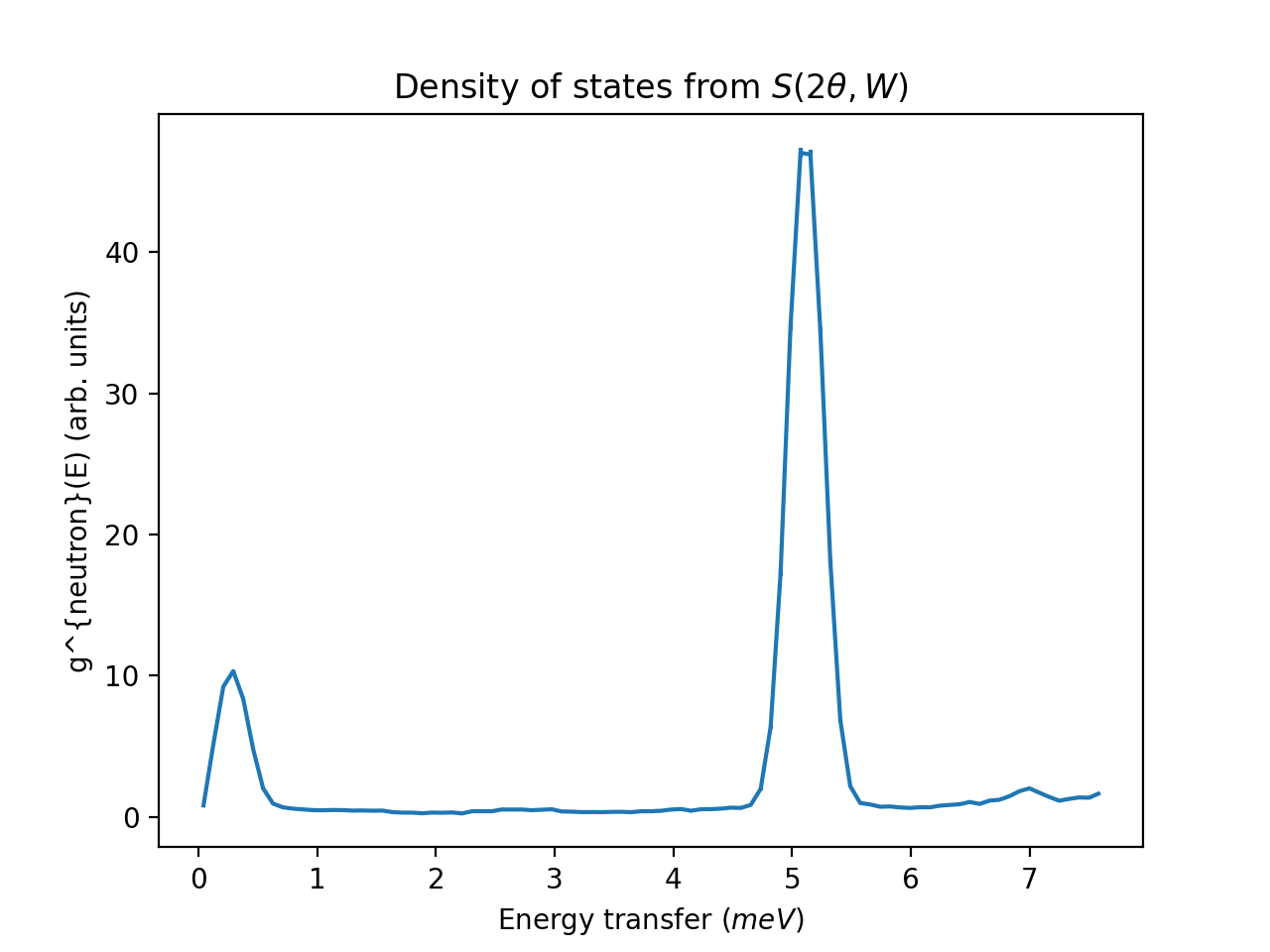
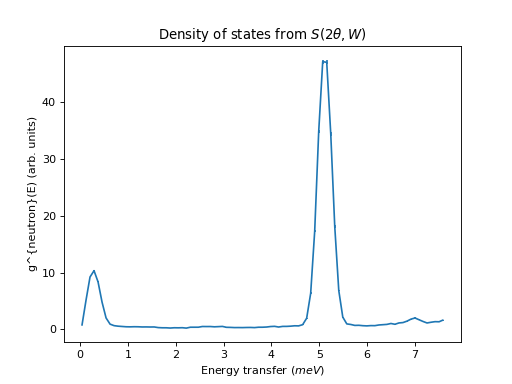
ILL Example using S(2theta, E) as input
from mantid import mtd
from mantid.simpleapi import *
import matplotlib.pyplot as plt
ws = DirectILLCollectData('ILL/IN4/087294.nxs')
DirectILLReduction(ws, OutputWorkspace='sqw', OutputSofThetaEnergyWorkspace='stw')
temperature = ws.run().getProperty('sample.temperature').value
dos = ComputeIncoherentDOS('stw', Temperature=temperature, EnergyBinning='0, Emax')
fig, axis = plt.subplots(subplot_kw={'projection':'mantid'})
axis.errorbar(dos)
axis.set_title('Density of states from $S(2\\theta,W)$')
# Uncomment the line below to show the plot.
#fig.show()
mtd.clear()
(Source code, png, hires.png, pdf)
{kind=link}
{kind=link}
Test Example
This example uses a generated dataset so that it will run on automated tests of the build system where the above datafiles do not exist.
ws = CreateSampleWorkspace(binWidth = 0.1, XMin = 0, XMax = 50, XUnit = 'DeltaE')
ws = ScaleX(ws, -25, "Add")
LoadInstrument(ws, InstrumentName='MARI', RewriteSpectraMap = True)
ws = SofQW(ws, [0, 0.05, 8], 'Direct', 25)
ws_DOS = ComputeIncoherentDOS(ws)
References#
Categories: AlgorithmIndex | Inelastic
Source#
Python: ComputeIncoherentDOS.py