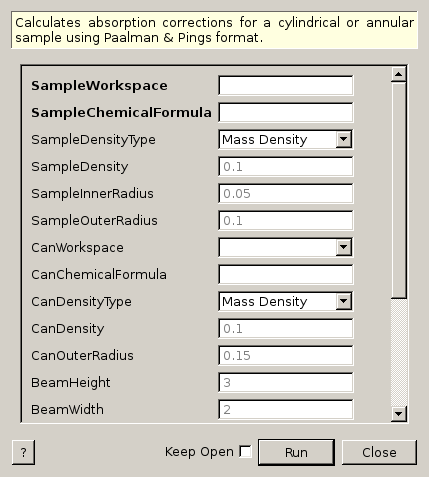
CylinderPaalmanPingsCorrection dialog.
Table of Contents
Calculates absorption corrections for a cylindrical or annular sample using Paalman & Pings format.
| Name | Direction | Type | Default | Description |
|---|---|---|---|---|
| SampleWorkspace | Input | MatrixWorkspace | Mandatory | Name for the input Sample workspace. |
| SampleChemicalFormula | Input | string | Mandatory | Sample chemical formula |
| SampleDensityType | Input | string | Mass Density | Use of Mass density or Number density. Allowed values: [‘Mass Density’, ‘Number Density’] |
| SampleDensity | Input | number | 0.1 | Mass density (g/cm^3) or Number density (atoms/Angstrom^3) |
| SampleInnerRadius | Input | number | 0.05 | Sample inner radius |
| SampleOuterRadius | Input | number | 0.1 | Sample outer radius |
| CanWorkspace | Input | MatrixWorkspace | Name for the input Can workspace. | |
| CanChemicalFormula | Input | string | Can chemical formula | |
| CanDensityType | Input | string | Mass Density | Use of Mass density or Number density. Allowed values: [‘Mass Density’, ‘Number Density’] |
| CanDensity | Input | number | 0.1 | Mass density (g/cm^3) or Number density (atoms/Angstrom^3) |
| CanOuterRadius | Input | number | 0.15 | Can outer radius |
| BeamHeight | Input | number | 3 | Beam height |
| BeamWidth | Input | number | 2 | Beam width |
| StepSize | Input | number | 0.002 | Step size |
| Interpolate | Input | boolean | True | Interpolate the correction workspaces to match the sample workspace |
| NumberWavelengths | Input | number | 10 | Number of wavelengths for calculation |
| Emode | Input | string | Elastic | Energy transfer mode. Allowed values: [‘Elastic’, ‘Indirect’, ‘Direct’, ‘Efixed’] |
| Efixed | Input | number | 0 | Analyser energy (mev). By default will be read from the instrument parameters. Specify manually to override. This is used in energy transfer modes other than Elastic. |
| OutputWorkspace | Output | WorkspaceGroup | Mandatory | The output corrections workspace group |
Calculates absorption corrections for a cylindrical or annular sample giving
output in the Paalman and Pings absorption factors: 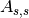 (correction
factor for scattering and absorption in sample),
(correction
factor for scattering and absorption in sample), 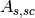 (scattering in
sample and absorption in sample and container),
(scattering in
sample and absorption in sample and container), 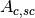 (scattering in
container and absorption in sample and container) and
(scattering in
container and absorption in sample and container) and 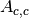 (scattering and absorption in container).
(scattering and absorption in container).
The input workspace must have a fully defined instrument.
The algorithm operates in different energy transfer modes, where the incident (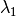 ) and the final (
) and the final (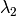 )
wavelengths are defined as follows:
)
wavelengths are defined as follows:
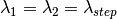
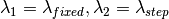
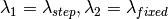
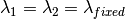 ,
,where 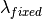 is computed from the Efixed value corresponding to the monochromator or the analyser, and
is computed from the Efixed value corresponding to the monochromator or the analyser, and
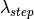 iterates equidistantly over the wavelength points in the input workspace x-axis, controlled by NumberWavelengths property.
iterates equidistantly over the wavelength points in the input workspace x-axis, controlled by NumberWavelengths property.
Therefore, in all the modes except Efixed, the input workspaces must have the x-axis unit of Wavelength. In all the modes except Elastic, Efixed value is needed. By default it will be attempted to be read from the instrument parameters, but can be overridden by the homonym property. In the Efixed mode the NumberWavelengths and Interpolate options will be ignored.
Example:
# Create a sample workspace
sample = CreateSampleWorkspace(NumBanks=1, BankPixelWidth=1,
XUnit='Wavelength',
XMin=6.8, XMax=7.9,
BinWidth=0.1)
# Copy and scale it to make a can workspace
can = CloneWorkspace(InputWorkspace=sample)
can = Scale(InputWorkspace=can, Factor=1.2)
# Calculate absorption corrections
corr = CylinderPaalmanPingsCorrection(SampleWorkspace=sample,
SampleChemicalFormula='H2-O',
SampleInnerRadius=0.05,
SampleOuterRadius=0.1,
CanWorkspace=can,
CanChemicalFormula='V',
CanOuterRadius=0.15,
BeamHeight=0.1,
BeamWidth=0.1,
StepSize=0.002,
Emode='Indirect',
Efixed=1.845)
print 'Correction workspaces: %s' % (', '.join(corr.getNames()))
Output:
Correction workspaces: corr_ass, corr_assc, corr_acsc, corr_acc