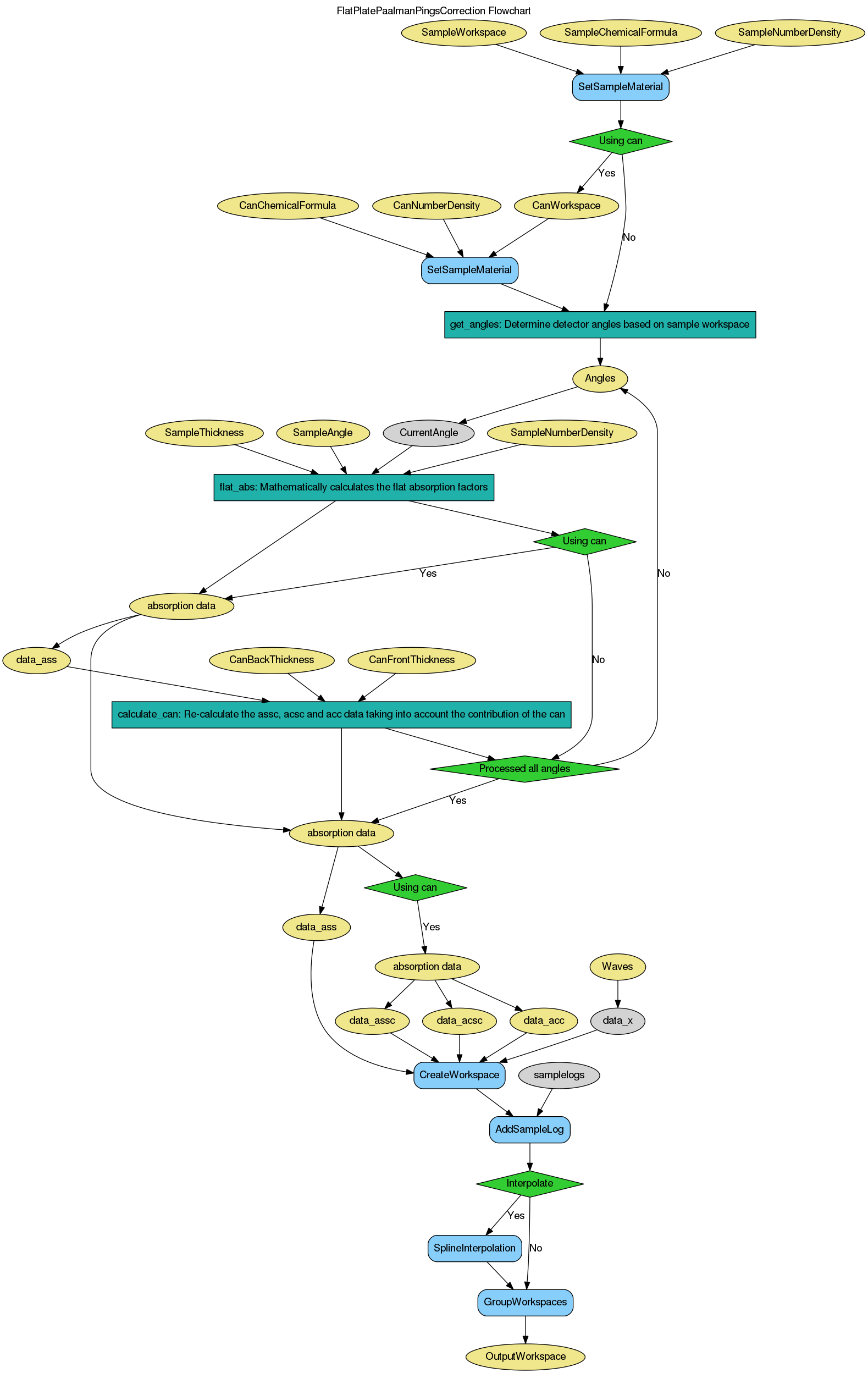
FlatPlatePaalmanPingsCorrection dialog.
Table of Contents
| Name | Direction | Type | Default | Description |
|---|---|---|---|---|
| SampleWorkspace | Input | MatrixWorkspace | Mandatory | Name for the input sample workspace |
| SampleChemicalFormula | Input | string | Mandatory | Sample chemical formula |
| SampleDensityType | Input | string | Mass Density | Use of Mass density or Number density. Allowed values: [‘Mass Density’, ‘Number Density’] |
| SampleDensity | Input | number | 0.1 | Mass density (g/cm^3) or Number density (atoms/Angstrom^3) |
| SampleThickness | Input | number | 0 | Sample thickness in cm |
| SampleAngle | Input | number | 0 | Angle between incident beam and normal to flat plate surface |
| CanWorkspace | Input | MatrixWorkspace | Name for the input container workspace | |
| CanChemicalFormula | Input | string | Container chemical formula | |
| CanDensityType | Input | string | Mass Density | Use of Mass density or Number density. Allowed values: [‘Mass Density’, ‘Number Density’] |
| CanDensity | Input | number | 0.1 | Mass density (g/cm^3) or Number density (atoms/Angstrom^3) |
| CanFrontThickness | Input | number | 0 | Container front thickness in cm |
| CanBackThickness | Input | number | 0 | Container back thickness in cm |
| NumberWavelengths | Input | number | 10 | Number of wavelengths for calculation |
| Interpolate | Input | boolean | True | Interpolate the correction workspaces to match the sample workspace |
| Emode | Input | string | Elastic | Energy transfer mode. Allowed values: [‘Elastic’, ‘Indirect’, ‘Direct’, ‘Efixed’] |
| Efixed | Input | number | 0 | Analyser energy (mev). By default will be read from the instrument parameters. Specify manually to override. This is used only in Efixed energy transfer mode. |
| OutputWorkspace | Output | WorkspaceGroup | Mandatory | The output corrections workspace group |
Calculates absorption corrections for an infinite flat plate sample giving output in the
Paalman and Pings absorption factors:
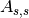 (scattering and absorption in sample),
(scattering and absorption in sample),
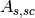 (scattering in sample and absorption in sample and container),
(scattering in sample and absorption in sample and container),
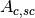 (scattering in container and absorption in sample and container)
and
(scattering in container and absorption in sample and container)
and 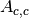 (scattering and absorption in container).
(scattering and absorption in container).
Details of the analytical method used to calculate the correction factors is available in RAL Technical Report 74-103.
Example:
# Create a sample workspace
sample = CreateSampleWorkspace(NumBanks=1, BankPixelWidth=1,
XUnit='Wavelength',
XMin=6.8, XMax=7.9,
BinWidth=0.1)
# Copy and scale it to make a can workspace
can = CloneWorkspace(InputWorkspace=sample)
can = Scale(InputWorkspace=can, Factor=1.2)
# Calculate absorption corrections
corr = FlatPlatePaalmanPingsCorrection(SampleWorkspace=sample,
SampleChemicalFormula='H2-O',
SampleThickness=0.1,
SampleAngle=45,
CanWorkspace=can,
CanChemicalFormula='V',
CanFrontThickness=0.01,
CanBackThickness=0.01,
Emode='Indirect',
Efixed=1.845)
print 'Correction workspaces: %s' % (', '.join(corr.getNames()))
Output:
Correction workspaces: corr_ass, corr_assc, corr_acsc, corr_acc