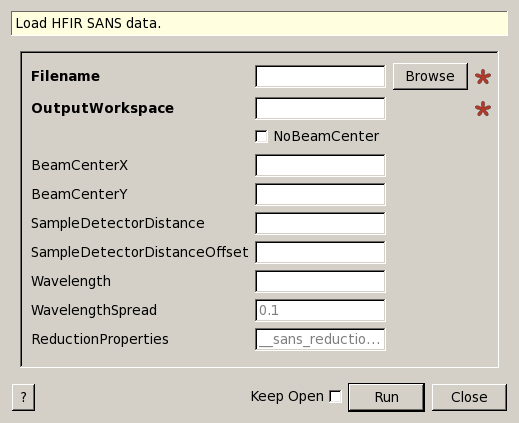
HFIRLoad dialog.
Table of Contents
| Name | Direction | Type | Default | Description |
|---|---|---|---|---|
| Filename | Input | string | Mandatory | The name of the input file to load. Allowed extensions: [‘.xml’] |
| OutputWorkspace | Output | MatrixWorkspace | Mandatory | Then name of the output workspace |
| NoBeamCenter | Input | boolean | False | If true, the detector will not be moved according to the beam center |
| BeamCenterX | Input | number | Optional | Beam position in X pixel coordinates |
| BeamCenterY | Input | number | Optional | Beam position in Y pixel coordinates |
| SampleDetectorDistance | Input | number | Optional | Sample to detector distance to use (overrides meta data), in mm |
| SampleDetectorDistanceOffset | Input | number | Optional | Offset to the sample to detector distance (use only when using the distance found in the meta data), in mm.Not used when SampleDetectorDistance is provided. |
| Wavelength | Input | number | Optional | Wavelength value to use when loading the data file (Angstrom). |
| WavelengthSpread | Input | number | 0.1 | Wavelength spread to use when loading the data file (default 0.0) |
| OutputMessage | Output | string | ||
| ReductionProperties | Input | string | __sans_reduction_properties |
Workflow algorithm that loads HFIR SANS data using the LoadSpice2D algorithm and applies basic corrections to the workspace. Those include:
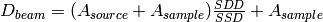
This algorithm is rarely called directly. It is called by HFIRSANSReduction, which will pass along the relevant beam center information through the ReductionProperties property.
Note
To run these usage examples please first download the usage data, and add these to your path. In MantidPlot this is done using Manage User Directories.
Example - Load a BioSANS data file:
workspace = HFIRLoad('BioSANS_empty_cell.xml')
r= mtd['workspace'].run()
print "SDD = %g" % r.getProperty('sample-detector-distance').value
Output:
SDD = 6000
Categories: Algorithms | Workflow\SANS\UsesPropertyManager