This function calculates the crystal field contribution to the molar magnetic susceptibility using the Van Vleck formula.
The function outputs the results by default in cgs units of cm3/mol == “emu/mol”.
There are also options to output the result in SI (m3/mol) or “atomic” units (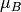 /Tesla/ion).
/Tesla/ion).
The magnetic susceptibility can be calculated by treating the magnetic field (Zeeman interaction) as a perturbation on the crystal field energy. To second order, the susceptibility per mole of magnetic ion is given by:
![\chi(T) = \frac{N_A}{Z} \sum_n \left[ \frac{| \langle V_n | g_J \mu_B \mathbf{J} | V_n \rangle | ^2}{k_B T}
- 2 \sum_{m \neq n} \frac{| \langle V_n | g_J \mu_B \mathbf{J} | V_m \rangle | ^2}{E_n - E_m} \right] \exp(-\beta E_n)](../_images/math/e31e383af9998fc405f2f97814815c9d455160d5.png)
where 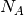 is Avogadro’s constant,
is Avogadro’s constant, 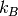 is Boltzmann’s constant,
is Boltzmann’s constant, 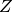 is the partition sum, and
is the partition sum, and
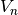 and
and 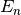 are the n-th wavefunction (eigenvector) and energy level (eigenvalue) of the unperturbed
crystal field Hamiltonian.
are the n-th wavefunction (eigenvector) and energy level (eigenvalue) of the unperturbed
crystal field Hamiltonian. 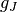 is the Landé g-factor, is the Bohr magneton and the moment operator
is defined as
is the Landé g-factor, is the Bohr magneton and the moment operator
is defined as 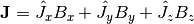 where
where 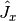 ,
, 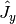 ,
and
,
and 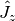 are the angular momentum operators in Cartesian coordinates, with
are the angular momentum operators in Cartesian coordinates, with  defined to
be along the quantisation axis of the crystal field (which is usually defined to be the highest symmetry rotation axis).
defined to
be along the quantisation axis of the crystal field (which is usually defined to be the highest symmetry rotation axis).
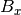 ,
, 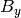 , and
, and 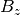 are the components of the unit vector pointing in the direction of the applied
magnetic field in this coordinate system.
are the components of the unit vector pointing in the direction of the applied
magnetic field in this coordinate system.
Finally, in order to account for the effect of any exchange interactions in the system which will shift the susceptibility curve up or down (analogous to the Curie-Weiss temperature), the actual magnetic susceptibility calculated by this function is:
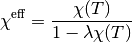
where 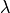 parameterises an effective exchange interaction and
parameterises an effective exchange interaction and 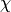 is the bare (paramagnetic Crystal Field)
susceptibility. A negative indicates overall antiferromagnetic interactions, whilst a positive
corresponds to overall ferromagnetic interactions.
is the bare (paramagnetic Crystal Field)
susceptibility. A negative indicates overall antiferromagnetic interactions, whilst a positive
corresponds to overall ferromagnetic interactions.
Here is an example of how to the crystal field parameters to a susceptibility dataset. All parameters disallowed by symmetry are fixed automatically. The “data” here is generated from the function itself, for a field along the [111] direction with respects to the crystal field parameters (not necessarily the [111] crystallographic direction).
The x-axis is given in Kelvin, and the susceptibility (y-axis) is in cgs units of cm3/mol (==emu/mol).
import numpy as np
# Build a reference data set
fun = 'name=CrystalFieldSusceptibility,Ion=Ce,B20=0.37737,B22=0.039770,B40=-0.031787,B42=-0.11611,B44=-0.12544,'
fun += 'Hdir=(1,1,1), Unit=cgs, inverse=1,'
# This creates a (empty) workspace to use with EvaluateFunction
x = np.linspace(1, 300, 300)
y = x * 0
e = y + 1
ws = CreateWorkspace(x, y, e)
# The calculated data will be in 'data', WorkspaceIndex=1
EvaluateFunction(fun, ws, OutputWorkspace='data')
# Change parameters slightly and fit to the reference data
fun = 'name=CrystalFieldSusceptibility,Ion=Ce,Symmetry=C2v,B20=0.4,B22=0.04,B40=-0.03,B42=-0.1,B44=-0.1,'
fun += 'Hdir=(1,1,1), Unit=cgs, inverse=1,'
fun += 'ties=(B60=0,B62=0,B64=0,B66=0,BmolX=0,BmolY=0,BmolZ=0,BextX=0,BextY=0,BextZ=0)'
# (set MaxIterations=0 to see the starting point)
Fit(fun, 'data', WorkspaceIndex=1, Output='fit',MaxIterations=100, CostFunction='Unweighted least squares')
# Using Unweighted least squares fit because the data has no errors.
# Extract fitted parameters
parws = mtd['fit_Parameters']
for i in range(parws.rowCount()):
row = parws.row(i)
if row['Value'] != 0:
print "%7s = % 7.5g" % (row['Name'], row['Value'])
Output (the numbers you see on your machine may vary):
B20 = 0.37737
B22 = 0.039788
B40 = -0.031787
B42 = -0.11611
B44 = -0.12544
Cost function value = 1.0921e-14
| Name | Type | Default | Description |
|---|---|---|---|
| Ion | String | Mandatory | An element name for a rare earth ion. Possible values are: Ce, Pr, Nd, Pm, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb. |
| Symmetry | String | C1 | A symbol for a symmetry group. Setting Symmetry automatically zeros and fixes all forbidden parameters. Possible values are: C1, Ci, C2, Cs, C2h, C2v, D2, D2h, C4, S4, C4h, D4, C4v, D2d, D4h, C3, S6, D3, C3v, D3d, C6, C3h, C6h, D6, C6v, D3h, D6h, T, Td, Th, O, Oh |
| powder | Boolean | false | Whether to calculate the powder averaged magnetisation or not. |
| Hdir | Vector | (0.,0.,1.) | The direction of the applied field w.r.t. the crystal field parameters |
| Unit | String | ‘bohr’ | The desired units of the output, either: ‘bohr’ (muB/T/ion), ‘SI’ (m^3/mol) or ‘cgs’ (cm^3/mol). |
| inverse | Boolean | false | Whether to output 1/chi(T) instead of chi(T). |
| Name | Default | Description |
|---|---|---|
| BmolX | 0.0 | The x-component of the molecular field. |
| BmolY | 0.0 | The y-component of the molecular field. |
| BmolZ | 0.0 | The z-component of the molecular field. |
| BextX | 0.0 | The x-component of the external field. |
| BextY | 0.0 | The y-component of the external field. |
| BextZ | 0.0 | The z-component of the external field. |
| B20 | 0.0 | Real part of the B20 field parameter. |
| B21 | 0.0 | Real part of the B21 field parameter. |
| B22 | 0.0 | Real part of the B22 field parameter. |
| B40 | 0.0 | Real part of the B40 field parameter. |
| B41 | 0.0 | Real part of the B41 field parameter. |
| B42 | 0.0 | Real part of the B42 field parameter. |
| B43 | 0.0 | Real part of the B43 field parameter. |
| B44 | 0.0 | Real part of the B44 field parameter. |
| B60 | 0.0 | Real part of the B60 field parameter. |
| B61 | 0.0 | Real part of the B61 field parameter. |
| B62 | 0.0 | Real part of the B62 field parameter. |
| B63 | 0.0 | Real part of the B63 field parameter. |
| B64 | 0.0 | Real part of the B64 field parameter. |
| B65 | 0.0 | Real part of the B65 field parameter. |
| B66 | 0.0 | Real part of the B66 field parameter. |
| IB21 | 0.0 | Imaginary part of the B21 field parameter. |
| IB22 | 0.0 | Imaginary part of the B22 field parameter. |
| IB41 | 0.0 | Imaginary part of the B41 field parameter. |
| IB42 | 0.0 | Imaginary part of the B42 field parameter. |
| IB43 | 0.0 | Imaginary part of the B43 field parameter. |
| IB44 | 0.0 | Imaginary part of the B44 field parameter. |
| IB61 | 0.0 | Imaginary part of the B61 field parameter. |
| IB62 | 0.0 | Imaginary part of the B62 field parameter. |
| IB63 | 0.0 | Imaginary part of the B63 field parameter. |
| IB64 | 0.0 | Imaginary part of the B64 field parameter. |
| IB65 | 0.0 | Imaginary part of the B65 field parameter. |
| IB66 | 0.0 | Imaginary part of the B66 field parameter. |
| Lambda | 0.0 | Effective exchange interaction |
Categories: FitFunctions | General