This document describes how crystal structure data can be processed and used in Mantid. For the understanding of the concepts of symmetry and space groups in Mantid it may be useful to read those introductory articles before proceeding with this document. While there is a short introduction into theoretical aspects this page is not a replacement for proper introductory text books on the subject where all these principles are explained in great detail and on a much more general basis.
A crystal is modelled as an infinitely repeating three-dimensional arrangement of scatterers, usually atoms. Due to the periodic nature of crystals, the entire object can be described by specifying the repeated unit and how it is repeated. These information are called “crystal structure” and comprise three components:
The description of the basis depends on the model’s level of detail. In the simplest case it could be a list of point scatterers that are fully characterized by three coordinates (x, y and z in terms of the lattice vectors) and a scattering length. In reality however, the scatterers are usually atoms that fluctuate about their average position due to thermal agitation. A basic way to model this motion is to assume it to be an isotropic phenomenon, allowing the description with one single parameter that quantifies the radius of a sphere in which the scatterer oscillates harmonically. This so called Debye-Waller-factor will be introduced later on.
Another important parameter for a basic description of the basis is the so called occupancy. It describes the fraction of the total number of scatterer-positions that is actually occupied. A common example where this is required are unordered binary metal mixtures where the same position in the crystal structure is partly filled with two different atoms in a randomly distributed manner.
To summarize, a very basic model of the basis comprises a list of scatterers that are in turn characterized by six parameters:
Knowledge of the space group makes it possible to derive all scatterers in the entire unit cell. The symmetry operations
of the space group map each scatterer-position onto a set of equivalent positions that are consequently occupied by the
same type of scatterer as well. Since the unit cell is repeated infinitely in all three directions of space, it is
enough to describe one unit cell. Finally, the lattice is described by six parameters as well, the lengths of the three
lattice vectors (usually given in 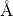 ) and the three angles (in degree) between these vectors.
) and the three angles (in degree) between these vectors.
In a diffraction experiment the periodic arrangement of atoms is probed using radiation, in this case in the form of
neutrons, of an appropriate wavelength (on the same scale of interatomic distances, typically between 0.5 and
5 ). The incident beam interacts with the scatterers and in certain orientations the beam is
“reflected” by a flock of lattice planes, a phenomenon which is described by Bragg’s law:
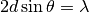
In this equation :math: d is the spacing between the lattice planes, 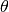 is the angle of the incoming beam
and the lattice plane normal and lambda is the wavelength of the radiation. In an experiment theta and lambda are
usually limited, thus they are limiting the range of interplanar spacings that can be probed. In Bragg’s law the lattice
plane families are only described by one parameter, the interplanar distance. But each lattice plane family also has an
orientation in space which can be described by the plane’s normal vector. Usually the vector is given in terms of the
reciprocal lattice of the structure, where it is reduced to three integers H, K, L, the so called Miller indices. With
knowledge of the unit cell (and thus the
is the angle of the incoming beam
and the lattice plane normal and lambda is the wavelength of the radiation. In an experiment theta and lambda are
usually limited, thus they are limiting the range of interplanar spacings that can be probed. In Bragg’s law the lattice
plane families are only described by one parameter, the interplanar distance. But each lattice plane family also has an
orientation in space which can be described by the plane’s normal vector. Usually the vector is given in terms of the
reciprocal lattice of the structure, where it is reduced to three integers H, K, L, the so called Miller indices. With
knowledge of the unit cell (and thus the 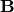 -matrix), the interplanar spacing can also
be computed like this:
-matrix), the interplanar spacing can also
be computed like this:
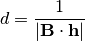
The parameters taken into account so far determine the geometric characteristics of Bragg-reflections, i.e. their position on a detector and their time of flight. But besides these, each reflection also has an intensity. The intensity is proportional to the squared structure factor, which depends on the kind and arrangement of scatterers in the unit cell. The structure factor is a complex number and can be calculated for a certain HKL by summing the contributions of all N atoms j in the unit cell:
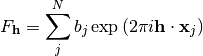
In the above equation 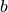 is the scattering length,
is the scattering length, 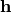 is the Miller index triplet HKL and
is the Miller index triplet HKL and
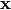 contains the fractional coordinates of the j-th atom. To take into account isotropic thermal
motion of atoms, the term is multiplied with the Debye-Waller factor:
contains the fractional coordinates of the j-th atom. To take into account isotropic thermal
motion of atoms, the term is multiplied with the Debye-Waller factor:
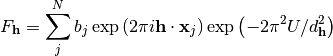
Here, 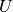 is the isotropic atomic displacement parameter, usually given in
is the isotropic atomic displacement parameter, usually given in 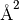 and
and
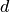 is the lattice spacing discussed above. There are other, more complex models to describe the movement of
atoms, taking into account anisotropic movement and also anharmonic effects.
is the lattice spacing discussed above. There are other, more complex models to describe the movement of
atoms, taking into account anisotropic movement and also anharmonic effects.
The concepts described above are available through the Python interface of Mantid. Crystal structures are represented by a class that stores the three necessary pieces of information. Objects of that class can be created by supplying string representations of those three arguments.
from mantid.geometry import CrystalStructure
silicon = CrystalStructure("5.431 5.431 5.431", "F d -3 m", "Si 0 0 0 1.0 0.05")
unitCell = silicon.getUnitCell()
print('Crystal structure of silicon:')
print(' Unit cell: {0} {1} {2} {3} {4} {5}'.format(unitCell.a(), unitCell.b(), unitCell.c(), unitCell.alpha(), unitCell.beta(), unitCell.gamma()))
spaceGroup = silicon.getSpaceGroup()
print(' Space group: {0}'.format(spaceGroup.getHMSymbol()))
print(' Point group: {0}'.format(spaceGroup.getPointGroup().getHMSymbol()))
scatterers = silicon.getScatterers()
print(' Total number of scatterers: {0}'.format(len(scatterers)))
for i, scatterer in enumerate(scatterers):
print(' {0}: {1}'.format(i,scatterer))
The above script produces the following output:
Crystal structure of silicon:
Unit cell: 5.431 5.431 5.431 90.0 90.0 90.0
Space group: F d -3 m
Point group: m-3m
Total number of scatterers: 1
0: Si 0 0 0 1 0.05
In general, the unit cell must be specified using either 3 or 6 space-separated floating point numbers, representing
the three axis lengths and the three angles between them. The list of scatterers is required to be a semi-colon
separated list of strings which contain the following information: Element symbol, x, y, z (fractional coordinates),
occupancy (between 0 and 1) and isotropic atomic displacement parameter. The fractional coordinates can also be given
as fractions (for example 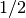 or
or 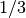 ) and for giving the coordinates in hexagonal or trigonal structures
this is highly recommended as there may be precision problems with decimal numbers.
) and for giving the coordinates in hexagonal or trigonal structures
this is highly recommended as there may be precision problems with decimal numbers.
While the CrystalStructure class is storing information, there is another class that makes use of these information to generate reflections and calculate structure factors. This class is called ReflectionGenerator and can be constructed from a CrystalStructure-object:
from mantid.geometry import CrystalStructure, ReflectionGenerator
from mantid.kernel import V3D
silicon = CrystalStructure("5.431 5.431 5.431", "F d -3 m", "Si 0 0 0 1.0 0.05")
generator = ReflectionGenerator(silicon)
# Create list of unique reflections between 0.7 and 3.0 Angstrom
hkls = generator.getUniqueHKLs(0.7, 3.0)
print('There are {} unique reflections for Si in the specified resolution range.'.format(len(hkls)))
print('The reflection [222] is' + (' not' if not V3D(2, 2, 2) in hkls else '') + ' contained in the list.')
There are 20 unique reflections for Si in the specified resolution range.
The reflection [222] is contained in the list.
Checking the reflection conditions of space group 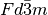 (origin choice 1) in the International Tables for
Crystallography shows that if an atom is on the 8a position, additional conditions apply (
(origin choice 1) in the International Tables for
Crystallography shows that if an atom is on the 8a position, additional conditions apply (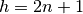 or
or
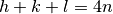 for general reflections). Using these additional conditions, the 222 reflection should in fact
not be in the list. This can be verified by calculating structure factors for the list of reflections and check if
there are very small values present.
for general reflections). Using these additional conditions, the 222 reflection should in fact
not be in the list. This can be verified by calculating structure factors for the list of reflections and check if
there are very small values present.
from mantid.geometry import CrystalStructure, ReflectionGenerator
import numpy as np
silicon = CrystalStructure("5.431 5.431 5.431", "F d -3 m", "Si 0 0 0 1.0 0.05")
generator = ReflectionGenerator(silicon)
# Create list of unique reflections between 0.7 and 3.0 Angstrom
hkls = generator.getUniqueHKLs(0.7, 3.0)
# Calculate structure factors for those HKLs
fSquared = generator.getFsSquared(hkls)
# Find HKLs with very small structure factors:
zeroFSquared = [(hkl, sf) for hkl, sf in zip(hkls, fSquared) if sf < 1e-9]
print('HKL F^2')
for hkl, sf in zeroFSquared:
print ('{0} {1}'.format(hkl, np.round(sf, 2)))
The output of the above script should show three reflections with very small values for 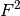 . Their indices
violate the special conditions mentioned in the previous paragraph, so the reflections are actually extinct:
. Their indices
violate the special conditions mentioned in the previous paragraph, so the reflections are actually extinct:
HKL F^2
[2,2,2] 0.0
[4,4,2] 0.0
[6,2,2] 0.0
Those three reflections are included in the list of unique HKLs, because the standard method to determine whether a reflection is allowed or not uses the space group symmetry which only reflects the general conditions listed in ITA. It is however possible to exclude those reflections at the cost of more computations by making use of the structure factor calculation. This can either be done by passing an additional enum-value of the type ReflectionConditionFilter to the constructor of ReflectionGenerator or by passing it to the actual generator function:
from mantid.geometry import CrystalStructure, ReflectionGenerator, ReflectionConditionFilter
from mantid.kernel import V3D
silicon = CrystalStructure("5.431 5.431 5.431", "F d -3 m", "Si 0 0 0 1.0 0.05")
generator = ReflectionGenerator(silicon)
# Create list of unique reflections between 0.7 and 3.0 Angstrom, use structure factors for filtering
hkls = generator.getUniqueHKLsUsingFilter(0.7, 3.0, ReflectionConditionFilter.StructureFactor)
print('There are {} unique reflections for Si in the specified resolution range.'.format(len(hkls)))
print('The reflection [222] is' + (' not' if not V3D(2, 2, 2) in hkls else '') + ' contained in the list.')
With this option, the three reflections from the example above are missing and as an indicator, the [222] reflection is actually checked:
There are 17 unique reflections for Si in the specified resolution range.
The reflection [222] is not contained in the list.
Other options for filtering are Centering and None. If the latter one is used the reflections are only filtered
according to their -value to fit the specified range.
Another capability of ReflectionGenerator is the calculation of -values for a list of HKLs, very similar
to the process for :
from mantid.geometry import CrystalStructure, ReflectionGenerator, ReflectionConditionFilter
import numpy as np
silicon = CrystalStructure("5.431 5.431 5.431", "F d -3 m", "Si 0 0 0 1.0 0.05")
generator = ReflectionGenerator(silicon)
# Create list of unique reflections between 0.7 and 3.0 Angstrom
hkls = generator.getUniqueHKLsUsingFilter(0.7, 3.0, ReflectionConditionFilter.StructureFactor)
# Calculate d and F^2
dValues = generator.getDValues(hkls)
fSquared = generator.getFsSquared(hkls)
pg = silicon.getSpaceGroup().getPointGroup()
# Make list of tuples and sort by d-values, descending, include point group for multiplicity.
reflections = sorted([(hkl, d, fsq, len(pg.getEquivalents(hkl))) for hkl, d, fsq in zip(hkls, dValues, fSquared)],
key=lambda x: x[1] - x[0][0]*1e-6, reverse=True)
print('{0:<8}{1:>8}{2:>8}{3:>4}'.format('HKL', 'd', 'F^2', 'M'))
for reflection in reflections:
print('{0!s:<8}{1:>8.5f}{2:>8.2f}{3:>4}'.format(*reflection))
This script will print a table with the reflections including their -value, and multiplicity due to point group
symmetry:
HKL d F^2 M
[2,2,0] 1.92015 645.02 12
[3,1,1] 1.63751 263.85 24
[4,0,0] 1.35775 377.63 6
[3,3,1] 1.24596 154.47 24
[4,2,2] 1.10860 221.08 24
[3,3,3] 1.04520 90.43 8
[5,1,1] 1.04520 90.43 24
[4,4,0] 0.96007 129.43 12
[5,3,1] 0.91801 52.94 48
[6,2,0] 0.85872 75.78 24
[5,3,3] 0.82822 31.00 24
[4,4,4] 0.78390 44.36 8
[5,5,1] 0.76049 18.15 24
[7,1,1] 0.76049 18.15 24
[6,4,2] 0.72575 25.97 48
[5,5,3] 0.70706 10.62 24
[7,3,1] 0.70706 10.62 48
This concept page explains what’s available in the Python interface. Some underlying parts may be interesting for C++ developers, as the concepts of generating and filtering HKLs are pretty much hidden behind the ReflectionGenerator class in the Python interface. More detail is available in the generated C++ documentation.
Category: Concepts