\(\renewcommand\AA{\unicode{x212B}}\)
AngularAutoCorrelationsSingleAxis dialog.
Table of Contents
Calculates the angular auto-correlation of molecules in a simulation along a user-defined axis. The axis is defined by the vector connecting the average position of species two and the average position of species one (user input). Timestep must be specified in femtoseconds.
| Name | Direction | Type | Default | Description |
|---|---|---|---|---|
| InputFile | Input | string | Mandatory | Input .nc file with an MMTK trajectory |
| Timestep | Input | string | 1.0 | Time step between two coordinates in the trajectory, fs |
| SpeciesOne | Input | string | Specify the first species, e.g. H, He, Li… | |
| SpeciesTwo | Input | string | Specify the second species, e.g. H, He, Li… | |
| OutputWorkspace | Output | Workspace | Mandatory | Output workspace name |
| OutputWorkspaceFT | Output | Workspace | Mandatory | Fourier Transform output workspace name |
Loads a netcdf file generated by nMoldyn containing MMTK format trajectories. The algorithm calculates angular auto-correlations of molecule in the simulation along a user-defined axis. The trajectory file must therefore contain molecule definitions. The axis vector is drawn from the average position of atoms of type SpeciesOne to the average position of atoms of type SpeciesTwo.
output_ws, output_ws_ft = AngularAutoCorrelationsSingleAxis(InputFile = 'trajectory_methyliodide.nc',
Timestep = '10.0',
SpeciesOne = 'C',
SpeciesTwo = 'I')
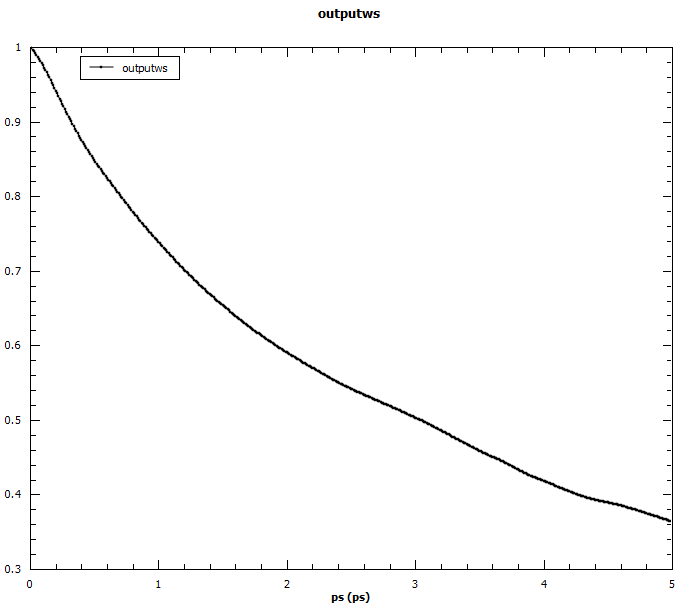
Angular auto-correlations calculated for methyliodide.
Categories: AlgorithmIndex | Simulation
Python: AngularAutoCorrelationsSingleAxis.py (last modified: 2020-03-27)