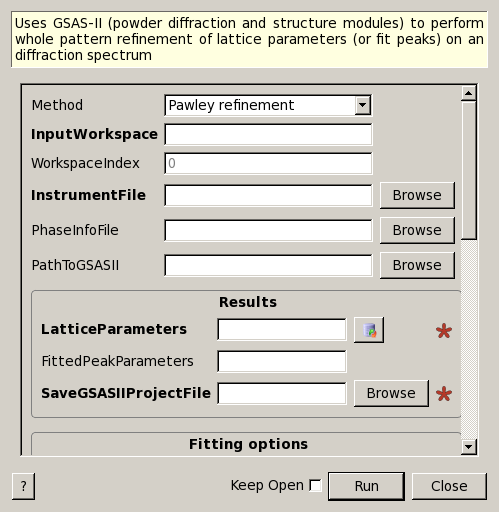
GSASIIRefineFitPeaks dialog.
Table of Contents
Uses GSAS-II (powder diffraction and structure modules) to perform whole pattern refinement of lattice parameters (or fit peaks) on an diffraction spectrum
| Name | Direction | Type | Default | Description |
|---|---|---|---|---|
| Method | Input | string | Pawley refinement | Rietveld corresponds to the Calculate/Refine option of the GSAS-II GUI. Peak fitting is single peak (does not use phase information and corresponds to the option Peaks List/Peak Fitting/PeakFitType of the GSAS-II GUI. The third alternative requires a list of peaks which can be bassed in the properties ExpectedPeaks and ExpectedPeaksFromFile. Allowed values: [‘Pawley refinement’, ‘Rietveld refinement’, ‘Peak fitting’] |
| InputWorkspace | Input | MatrixWorkspace | Mandatory | Workspace with spectra to fit peaks. ToF is expected X unit. |
| WorkspaceIndex | Input | number | 0 | Index of the workspace for the spectrum to fit. By default the first spectrum will be processed (that is, the only spectrum for focussed data workspaces. |
| InstrumentFile | Input | string | Mandatory | File with instrument parameters (in GSAS format). Allowed extensions: [‘.par’, ‘.prm’, ‘.ipar’, ‘.iparm’] |
| PhaseInfoFile | Input | string | File with phase information for the material. Allowed extensions: [‘.cif’] | |
| PathToGSASII | Input | string | Optional path to GSAS-II software installation. This will be used to import several Python modules from GSAS-II. | |
| GoF | Output | number | Goodness of fit value (Chi squared). | |
| Rwp | Output | number | Weighted profile R-factor (Rwp) discrepancy index for the goodness of fit. | |
| LatticeParameters | Output | TableWorkspace | Mandatory | Table to output the the lattice parameters (refined). |
| FittedPeakParameters | Input | string | Name for an (output) table of fitted parameters. This is used with the peak fitting method. The table will have one row per peak found. | |
| SaveGSASIIProjectFile | Input | string | Mandatory | GSAS-II project file (that can be openened in the GSAS-II GUI). Allowed extensions: [‘.gpx’] |
| BackgroundType | Input | string | Chebyshev | Type of background for the peak fitting. Currently only the default option of GSAS-II (chebyshev) is supported. Allowed values: [‘Chebyshev’, ‘None’] |
| MinX | Input | number | Optional | Minimum x value for the fitting, in the same units as the input workspace (TOF). Defines the range or domain of fitting together with the property MaxX. Leave empty to use the whole range |
| MaxX | Input | number | Optional | Maximum x value for the fitting, in the same units as the input workspace (TOF). Defines the range or domain of fitting together with the property MinX. Leave empty to use the whole range |
| PawleyDmin | Input | number | 1 | For Pawley refiment: as defined in GSAS-II, the minimum d-spacing to be used in a Pawley refinement. Please refer to the GSAS-II documentation for full details. |
| PawleyNegativeWeight | Input | number | 0 | For Pawley refinement: as defined in GSAS-II, the weight for a penalty function applied during a Pawley refinement on resulting negative intensities. Please refer to the GSAS-II documentation for full details. |
| ExpectedPeaks | Input | dbl list | A list of dSpacing values for the peak centers. These will be converted into TOF to find expected peaks. | |
| ExpectedPeaksFromFile | Input | string | Load from this file a list of dSpacing values to be converted into TOF . This takes precedence over ‘ExpectedPeaks’ when both options are given. Allowed extensions: [‘.csv’] | |
| RefineCenter | Input | boolean | False | Whether to refine the peak centers. |
| RefineIntensity | Input | boolean | False | Whether to refine the peak function intensity parameters (assuming a shape of type back-to-back exponential convoluted with pseudo-voigt (BackToBackExponentialPV). |
| RefineAlpha | Input | boolean | False | Whether to refine the peak function beta parameters (assuming a BackToBackExponentialPV peak shape. |
| RefineBeta | Input | boolean | False | Whether to refine the peak function beta parameters (assuming a BackToBackExponentialPV peak shape. |
| RefineSigma | Input | boolean | True | Whether to refine the peak function sigma parameters (assuming a BackToBackExponentialPV peak shape. |
| RefineGamma | Input | boolean | True | Whether to refine the peak function gamma parameters (assuming a BackToBackExponentialPV peak shape. |
Warning
This algorithm is experimental and at the moment is being developed for a specific technique. It might be changed, renamed or even removed without a notification, should instrument scientists decide to do so.
Warning
This algorithm requires GSAS-II, with custom modifications to enable it to be used from Mantid. Please contact the Mantid developers for details. The GSAS-II installation instructions are available from the GSAS-II website.
Uses GSAS-II [TobyVonDreele2013] as external software to fit peaks to a powder / engineering diffraction pattern. Here the process of peak fitting is in the context of Rietveld / Pawley / Le Bail analysis [LeBail2005]
The algorithm supports three refinement or fitting methods: Pawley refinement, Rietveld refinement, and single peak fitting (or “Peaks List” of GSAS-II). The first two methods of this algorithm implement whole diffraction pattern fitting whereas the third method fits peaks individually. The use of this algorithm is very close to the examples described in these two GSAS-II tutorials: Rietveld fitting / CW Neutron Powder fit for Yttrium-Iron Garnet, and Getting started / Fitting individual peaks & autoindexing, The functionality of this algorithm is based on the powder calculation module and the structure routines of GSAS-II.
To run this algorithm GSAS-II must be installed and it must be available for importing from the Mantid Python interpreter. This algorithm requires a modified version of GSAS-II. Please contact the developers for details.
The methods “Pawley refinement” and “Rietveld refinement” of this algorithm are equivalent to the function “Calculate / Refine” from the main menu of the GSAS-II GUI. The method “Peak fitting” is equivalent to the “Peak Fitting / Peak fit” action of the “Peaks List” window menu of the GSAS-II GUI.
The main inputs required are histogram data, an instrument definition parameter (in GSAS format, readable by GSAS-II), and various parameters for the fitting/refinement process. Phase information is also required to use the Pawley and Rietveld refinement.
The phase information must be provided in CIF format
(Crystallographic Information File).
When phase information is available and the Rietveld/Pawley method is
used the algorithm will output the lattice parameters in a table
workspace. The values are given for the the full set of lattice
parameters (three lattice constants, three angles, and volume in this
sequence: a, b, c, alpha, beta, gamma, volume). The a,b, and c values
are given in Angstroms (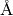 ). The angles are given
in degrees, and the volume in
). The angles are given
in degrees, and the volume in 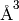 .
.
The algorithm provides goodness-of-fit estimates in the outputs GoF and Rwp or weighted profile R-factor [Toby2008]. The Rwp is given as a percentage value.
Note that the option to save the GSAS-II project file (SaveGSASIIProjectFile) is mandatory. This is a requirement of GSAS-II. These project files can be opened in the GSAS-II GUI for further processing and analysis of the data.
When Pawley refinement is selected as refinement method the flag for histogram scale factor refinement is disabled, as recommended in the GSAS-II documentation, as this cannot be refined simultaenously with the Pawley reflection intensities.
The GSAS-II Rietveld/Pawley refinement process writes lattice parameters and extensive additional information in an output file with the same name as the output GSAS-II project file but with extension ”.lst”. This is noted in a log message that specifies where the file has been written (next to the output project file).
When fitting individual peaks using the peak fitting method (not using Rietveld/Pawley refinement), the algorithm only supports peaks with shape of type back-to-back exponential convoluted with pseudo-voigt (BackToBackExponentialPV). It is possible to enable the refinement of the different function parameters via several properties (RefineAlpha, RefineSigma, etc.). The fitted peak parameters are given in an output table with as many rows as peaks have been found. The columns of the table give the parameters fitted, similarly to the information found in the “Peaks List” window of the GSAS-II GUI. These results are printed in the log messages as well.
For fitting single peaks, one at a time, see also EnggFitPeaks. For other algorithms that implement different variants of whole diffraction pattern refinement and fitting see also PawleyFit and LeBailFit.
References:
| [LeBail2005] | Le Bail, A (2005). “Whole Powder Pattern Decomposition Methods and Applications: A Retrospection”. Powder Diffraction 20(4): 316-326. |
| [TobyVonDreele2013] | Toby, B. H., & Von Dreele, R. B. (2013). “GSAS-II: the genesis of a modern open-source all purpose crystallography software package”. Journal of Applied Crystallography, 46(2), 544-549. |
| [Toby2008] | Toby, B. H. (2008). “R factors in Rietveld analysis: How good is good enough?”. Powder Diffraction, 21(1), 67-70. |
Example - Pawley refinement of lattice parameters from a diffraction spectrum
# You would normally generate the focused file using the Engg GUI or,
# alternatively, with commands like these:
#
# wks = Load('ENGINX00256663-256675')
# wks_ceria = Load('ENGINX00255924')
# wks_vana = Load('ENGINX00254854')
# # Using default expected peaks for Ceria
# difa, difc, tzero, peaks_tbl = EnggCalibrate(InputWorkspace=wks_ceria, VanadiumWorkspace=wks_vana, Bank='North')
# wks_focused = EnggFocus(InputWorkspace=wks, VanadiumWorkspace=wks_vana, Bank='North')
# SaveNexus(InputWorkspace=wks_focused, Filename='focused_bank1_ENGINX00256663.nxs')
#
wks=Load('focused_bank1_ENGINX00256663.nxs')
GoF, Rwp, lattice_tbl = GSASIIRefineFitPeaks(InputWorkspace=wks,
InstrumentFile='ENGINX_255924_254854_North_bank.par',
PhaseInfoFile='FE_ALPHA.cif',
PathToGSASII='/home/user/gsas',
SaveGSASIIProjectFile='example_gsas2_project',
LatticeParameters='lattice_tbl')
print "Goodness of fit coefficient: {0:.5f}".format(GoF)
print "Weighted profile R-factor (Rwp): {0:.5f}".format(Rwp)
print ("Lattice parameters, a: {a}, b: {b}, c: {c}, alpha: {alpha}, beta: {beta}, gamma: {gamma}, "
"Volume: {volume:.3f}".format(**lattice_tbl.row(0)))
Output:
Goodness of fit coefficient: 3.63591
Weighted profile R-factor (Rwp): 77.27831
Lattice parameters, a: 2.8665, b: 2.8665, c: 2.8665, alpha: 90.0, beta: 90.0, gamma: 90.0, Volume: 23.554
Example - Rietveld refinement of lattice parameters from a diffraction spectrum
wks=Load('focused_bank1_ENGINX00256663.nxs')
GoF, Rwp, lattice_tbl = GSASIIRefineFitPeaks(InputWorkspace=wks,
Method='Rietveld refinement',
InstrumentFile='ENGINX_255924_254854_North_bank.par',
PhaseInfoFile='FE_ALPHA.cif',
PathToGSASII='/home/user/gsas',
SaveGSASIIProjectFile='example_gsas2_project',
LatticeParameters='lattice_tbl')
print "Goodness of fit coefficient: {0:.5f}".format(GoF)
print "Weighted profile R-factor (Rwp): {0:.5f}".format(Rwp)
print ("Lattice parameters, a: {a}, b: {b}, c: {c}, alpha: {alpha}, beta: {beta}, gamma: {gamma}, "
"Volume: {volume:.3f}".format(**lattice_tbl.row(0)))
Output:
Goodness of fit coefficient: 3.62483
Weighted profile R-factor (Rwp): 77.03530
Lattice parameters, a: 2.8665, b: 2.8665, c: 2.8665, alpha: 90.0, beta: 90.0, gamma: 90.0, Volume: 23.554
Example - Fit several peaks from a diffraction spectrum
wks=Load('focused_bank1_ENGINX00256663.nxs')
params_tbl_name = 'tbl_fitted_params'
GoF, Rwp, lattice_tbl = GSASIIRefineFitPeaks(InputWorkspace=wks,
Method='Peak fitting',
FittedPeakParameters=params_tbl_name,
InstrumentFile='ENGINX_255924_254854_North_bank.par',
PhaseInfoFile='FE_ALPHA.cif',
PathToGSASII='/home/user/mantid-repos/gsas',
SaveGSASIIProjectFile='test_gsas2_project',
FittedPeakParameters=params_tbl_name)
tbl_fitted_params = mtd[params_tbl_name]
print "Fitted {0} peaks.".format(tbl_fitted_params.rowCount())
print ("Parameters of the first peak. Center: {Center:.6g}, intensity: {Intensity:.5f}, "
"alpha: {Alpha:.5f}, beta: {Beta:.5f}, sigma: {Sigma:.5f}, gamma: {Gamma:.5f}".
format(**tbl_fitted_params.row(0)))
Output:
Fitted 18 peaks.
Parameters of the first peak. Center: 38563.8, intensity: 26.22137, alpha: 0.13125, beta: 0.01990, sigma: 125475.11036, gamma: -6681.38965
Categories: Algorithms | Diffraction\Engineering | Diffraction\Fitting
Python: GSASIIRefineFitPeaks.py